
Legtöbbünknek a biológiai diverzitás hallatán a fajok sokfélesége jut eszébe, és nem véletlenül: manapság sok szó esik a mezőgazdasági vagy ipari tevékenységek biodiverzitást csökkentő hatásáról, a világszerte már kihalt és kihalással fenyegetett különböző élőlényekről. A kifejezés azonban nem csupán a fajok mennyiségét és egymáshoz viszonyított arányait takarja. A biológiai diverzitás különböző, ám egymásra épülő szinteken értelmezhető, ezek pedig a genetikai, faji és életközösségi szintű sokféleség. Mindezek közül leginkább a genetikai diverzitás tekinthető kulcsfontosságúnak, hiszen ez jelenti az evolúció „motorját”, a többi szint is ezen alapul.
A fajon vagy annak egy állományán belüli genetikai változatosság biztosíthatja, hogy az adott faj a hosszútávon drasztikusan megváltozó környezeti feltételek között se pusztuljon ki. Minél többfélék vagyunk, annál nagyobb rá az esély, hogy az élőhely megváltozása esetén is legalább néhányunk túlélő lesz, és tovább szaporodhat, biztosítva ezzel a teljes faj fennmaradását – ez az alapja magának a fajkeletkezésnek is.
A genetikai változatosságot különböző tényezők tartják fenn. A rekombináció jelensége során a sejtben lévő örökítőanyag átrendeződik az ivarsejtek kialakulásakor, ezáltal új kombinációk jönnek létre, frissítve a genetikai tartalmat. Horizontális géntranszfernek azt nevezzük, ha egy külső vektor, például vírus révén idegen dezoxiribonukleinsav (DNS) vagy ribonukleinsav (RNS) jut a szervezetbe. Baktériumok és vírusok esetében ez kifejezetten gyakori, de eukariótákban is előfordulhat. Tulajdonképpen ugyanez az eredménye a hibridizációnak is, amikor két külön fajhoz tartozó egyedek párosodása révén az utód mindkét faj genetikai jellemzőit hordozza.
Az említett jelenségek mellett a diverzitás igen nagymértékben a véletlen mutációknak is köszönhető. Minden élőlény minden egyes sejtosztódási folyamata során óriási energiamennyiség fordítódik a DNS-molekulák másolási hibáinak javítására. Az esetek elenyésző részében azonban a rendszer átsiklik egy-egy ilyen „hiba” felett, és ha az adott egyed ezt átörökíti az utódaira, azzal a populáció sokszínűségéhez járul hozzá. Újabb kártyát ad a paklihoz, amelyből a természetes szelekció húzhat, azaz „válogathat”. A hibázási lehetőségek száma elképzelhetetlenül nagy. Ha eltekintünk a kijavított vagy kifejezetten hátrányos, például betegségeket vagy pusztulást okozó változásoktól, még akkor is hatalmas mennyiségű észrevétlen mutáció maradhat az egymást követő generációk genetikai állományában, amelyek később egy-egy állomány és egyben a faj túlélésének zálogai is lehetnek.
A népszerű mitokondriális DNS
A XX. század második felében a genetikai kutatásokban nagy áttörést jelentett, amikor egy brit–amerikai kutatópáros, Frederick Sanger és Walter Gilbert kifejlesztette az örökítőanyag építőkockái, a nukleinsavak szekvenálásának módszerét. Ez lehetővé tette, hogy a DNS-molekulák apró eltérései felismerhetők, mi több, összehasonlíthatók legyenek. A populációgenetika, az evolúciókutatás sem menekülhetett az új módszerek alkalmazása révén felmerült lehetőségek elől, az állományok genetikai tartalmának feltérképezése ezt követően kezdődött el. Mára a laboratóriumi műveletek és a bioinformatikai, biostatisztikai eljárások sokat egyszerűsödtek, automatizálódtak az akkoriakhoz képest, így sok egyedtől származó, nagy mennyiségű adatot is viszonylag gyorsan és könnyen fel lehet dolgozni.

Az első lépés minden ilyen jellegű kutatásban az, hogy a kiválasztott élőlénycsoport egyedeiből mintákat szerezzünk be. Lehet ez egy levél, egy sejttenyészet, egy izomszövet darabkája vagy akár egy szőrcsomó, attól függően, hogy milyen faj képezi éppen a vizsgálatunk tárgyát vagy milyen lehetőségünk van a mintagyűjtés megvalósítására. Az összegyűjtött mintákból ezután ki kell vonni a genetikai információt tartalmazó molekulákat. Ehhez különböző módszereket dolgoztak ki, de mindnek az a lényege, hogy a DNS-molekulákat kiszabadítsuk a sejtekből, majd megtisztítsuk minden olyan anyagtól, ami a további laboratóriumi folyamatokat megzavarná vagy csökkentené a hatékonyságukat. Ezután rendelkezésünkre áll a kiválasztott élőlény „esszenciája”. A következő feladatunk a vizsgálathoz megfelelő nukleinsavszakasz kiválasztása annak tudatában, hogy pontosan mi is a célunk az adott vizsgálattal, milyen kérdésekre szeretnénk választ kapni.

Sok olyan gént és nem kódoló DNS-szakaszt térképeztek már fel és teszteltek világszerte, amelyek kiválóan alkalmasak a feladatra – ezeket nevezik genetikai markereknek. A mitokondrium DNS-molekulája az egyik olyan nukleinsav a sejtekben, amelyet széles körben használnak populációgenetikai, filogenetikai vizsgálatokhoz. Ennek oka, hogy a sejtmag DNS-éhez viszonyítva meglehetősen konzervatív, ami annyit tesz, hogy benne az utóbbihoz képest sokkal ritkábban következnek be a már említett mutációk. Népszerűségéhez az is hozzájárul, hogy kifejezetten kevés nem kódoló szakaszt tartalmaz, valamint, hogy – kevés kivételtől eltekintve – kizárólag anyai öröklődésű. A mitokondriális DNS-ét mindenki az anyjától örökli, így segítségével egyértelműen leírhatók az anya-utód kapcsolatok még akkor is, ha nagyon sok egyed mintáival dolgozunk.
A mitokondriális DNS mellett persze sok egyéb lehetőségünk is van. Gyakran használják például az Y-kromoszóma bizonyos szakaszait vagy akár egyéb, a testi kromoszómákon kódolódó enzimek génjeit. A kiválasztott DNS-szakaszról laboratóriumi körülmények között néhány óra leforgása alatt nagy mennyiségű másolatot készítünk a polimeráz láncreakció (PCR) nevű eljárás segítségével. Ezután következik a szekvenálás, azaz a DNS-molekula pontos szerkezetének, bázissorrendjének meghatározása.
Rokonsági kapcsolatok
A szekvenciáink most már készen állnak az elemzésre – „csak” össze kell hasonlítani őket. Hogy miként? Tegyük fel, hogy minden egyedtől rendelkezésünkre áll egy nagyjából ezer bázis hosszúságú DNS-szakasz szekvenciája. Ezek nem pontosan egyforma hosszúak, mert a szekvenálási folyamat nem indul és végződik minden minta DNS-szálunknak ugyanazon a pontján. A kapott szekvenciákat úgy tudjuk összeilleszteni, hogy az egymáshoz szerkezetileg leghasonlóbb szakaszaikat erre alkalmas szoftverek megkeresik, és ezek alapján egymás mellé rendezik őket. Miután az összeillesztett szekvenciák helyességét leellenőriztük, egyforma méretűekre vágjuk mindegyiket. Így már egyszerűen meghatározhatjuk – újabb számítógépes programok segítségével – azokat a pontokat a DNS-mintáinkban, ahol eltér egymástól a szerkezetük. Az ilyen polimorf helyek alapján kiderül, hogy a szekvenciáink hány olyan csoportba – haplotípusba – sorolhatók amelyeken belül a minták bázissorrendje megegyező. A haplotípusok számának, valamint az ezeket alkotó minták mennyiségének és egymáshoz viszonyított arányainak elemzésével állapítható meg a vizsgált populáció diverzitása, vagyis genetikai sokféleségének mértéke.
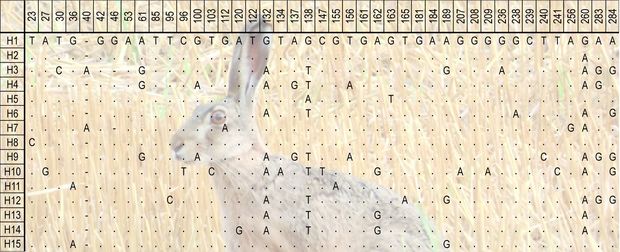
A polimorfizmusok mennyisége és a haplotípusok közötti hasonlóságok rávilágíthatnak a haplotípusok (illetve egyedek) rokonsági kapcsolataira is. Különböző modellek használatával evolúciós törzsfákat készíthetünk, melyeken ezek a rokonsági kapcsolatok szemmel láthatók. Nagyobb földrajzi terület egyedeit vizsgálva meghatározhatjuk a valódi populációk hozzávetőleges határait is, vagyis azt, hogy mely területek egyedei szaporodnak inkább egymás között, mint más területek egyedeivel. Az ilyen valódi szaporodási közösségek egymástól való elszeparálódásának idejére is fényt deríthetünk. Egyszerűen fogalmazva, meghatározhatjuk, hogy egy faj hazai és például bolgár populációi mikor különültek el egymástól a múltban.
Balkáni menedék
Jelenleg a mezei nyúllal (Lepus europaeus) kapcsolatban folytatunk egy nagy léptékű vizsgálatot a genetikai diverzitás témakörében a Debreceni Egyetemen, a Bolyai János Kutatási Ösztöndíj támogatásával. Fő kérdéseink, hogy a Kárpát-medencében élő állományok mikor és főként honnan vándoroltak ide az utolsó eljegesedés időszakát követően, valamint hogy mekkora a különbség – ha egyáltalán van – az itt élő és Európa más tájait benépesítő nyúlpopulációk között. Négy különböző DNS-szakasszal, vagyis markerrel dolgozunk, melyek közül kettő a mitokondriális DNS-ben található, kettő pedig a bőr- és szőrzetszínezet kialakításáért felelős nukleinsav szakasz a sejtmagban. Mintáink Európa különböző országaiból és Grúziából származnak, így lehetőségünk van az ilyen nagy földrajzi távolságokból adódó eltérések feltérképezésére.
Eddigi eredményeink, legalábbis ami a mitokondriális markereket illeti, azt mutatják, hogy a Kárpát-medencei nyulak a Balkán irányából vándoroltak jelenlegi élőhelyeikre, egy, az utolsó glaciális idején ott fennmaradó refúgiumból, azaz menedék területről. A populációk diverzitási értékei ugyanis délről északi irányba haladva fokozatosan csökkennek Európában. Ez megegyezik más, korábban Görögország és Bulgária területén végzett vizsgálatok adataival, amit azért érdemes megemlíteni, mert bizonyos kutatók úgy vélik, hogy a mezei nyúl számára több ilyen terület is létezhetett a délkelet-európai mellett Olaszországban és akár az Ibériai-félszigeten is. Egyelőre úgy tűnik, hogy nincs egyetértés a témában.
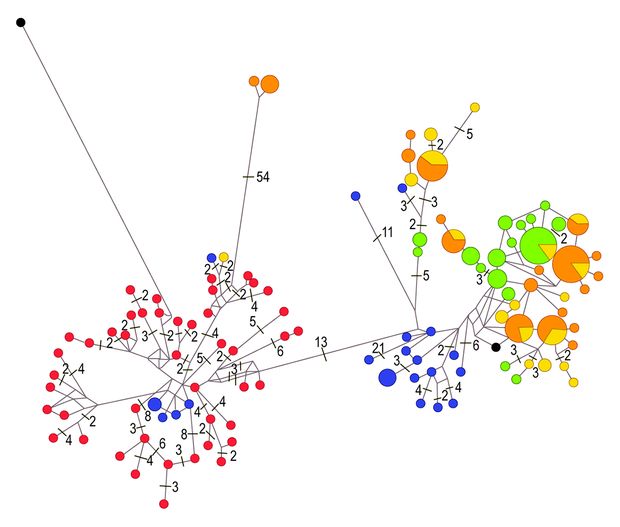
(A SZERZŐK SAJÁT ÁBRÁJA)
Adataink azt mutatják, hogy egyértelműen a grúziai állomány áll a legtávolabbi rokonságban az összes többi vizsgált populációval, és illeszkednek a korábban más kutatók által leírt eredményekhez, melyek szerint a mezei nyúl kontinensünkön két nagy csoportra – úgynevezett kládra – osztható. A keleti kládot kelet-balkáni, kelet-görögországi, anatóliai és további, izraeli nyúlpopulációk alkotják. A nyugati csoportba sorolható minden, az előbbi területektől nyugatra élő állomány. A két klád között minimális keveredés figyelhető meg még az elterjedési területeik átfedő zónájában is.
Emellett meg kell jegyezni, hogy kifejezetten nagymértékű hasonlóságot találtunk a magyarországi és olaszországi állományok között. Ez a genetikai és vadászati szakirodalmi adatokkal egybehangzó eredmény azt bizonyítja, hogy a XX. században indult, és a mai napig is tartó élő nyúl áttelepítési programok – melyek a túlvadászott olasz területek populációinak frissítését célozták – jelentősen csökkentik a faj európai diverzitását.
Mi a haszna?
Felmerülhet a kérdés, hogy ezekre a vizsgálatokra egyáltalán miért van szükség, miért hasznos ez nekünk. A kutatás talán legfontosabb tényezőjén, a kíváncsiságon túl természetesen gyakorlati haszna is van. Az élőlények genetikai hátterének megismerése tisztázhat rendszertani kérdéseket, vagy például képessé tehet arra, hogy megjósoljuk: egy-egy faj miképpen reagálna bizonyos környezeti változásokra a jövőben. Esetleg épp ellenkezőleg, múltbeli eseményekre deríthetünk fényt, meghatározhatjuk korábbi elterjedési területét, jobban megismerhetjük őseit. Az adatok iránymutatást nyújtanak további esetleges áttelepítési – frissítési programok megtervezéséhez, így elkerülhetjük a vadon élő állat- és növényfajok genetikai diverzitásának további csökkenését, ha a forráspopulációkat megfelelően választjuk ki. Ez alapján a módszer lehetőséget ad arra, hogy nekik megfelelően kezeljük a fajokat és élőhelyeiket annak érdekében, hogy a lehető legtovább élhessünk velük együtt ezen a bolygón.
SOÓS NOÉMI
KUSZA SZILVIA
2017/31

